 GS-US-540-5774(SIMPLE-Moderate)試験(国際共同第Ⅲ相試験)
GS-US-540-5774(SIMPLE-Moderate)試験(国際共同第Ⅲ相試験)
「禁忌を含む注意事項等情報」等はDIをご参照ください。
GS-US-540-5774(SIMPLE-Moderate)試験(国際共同第Ⅲ相試験)
Spinner CD, et al.: JAMA 2020; 324(11): 1048-1057.
本試験はギリアド・サイエンシズ社より支援を受けています。著者にギリアド・サイエンシズ社より支援を受けている者が含まれます。
社内資料(GS-US-540-5774試験)(承認時評価資料)
試験概要
| 目的 | SARS-CoV-2による中等症感染症患者を対象に、ベクルリー5日間投与、10日間投与及び標準療法のみの有効性と安全性を検討する。 |
| 試験デザイン | 多施設共同、無作為化、非盲検、並行群間試験 |
| 対象 | 12歳以上18歳未満かつ体重40kg以上、及び18歳以上の中等症のSARS-CoV-2による感染症患者584例 |
| 主な選択基準 |
|
| 主な除外基準 |
|
| 試験方法 | 患者を、標準療法のみの群、及び標準治療に加えて、ベクルリーを初日に200mg、2日目以降は100mgを1日1回5日目まで静脈内投与する群と10日目まで静脈内投与する群に1:1:1の割合で無作為に割り付けた。 |
| 主要評価項目 (検証的な解析項目) |
無作為化後10日目に7点順序尺度で評価した臨床状態 |
| 副次評価項目 | 有害事象の発現状況(ベクルリー投与初日~最終投与の30日後) |
| 探索的評価項目 | 回復までの時間、部分修正後の回復までの時間、臨床的改善までの時間、1点以上の改善までの期間、酸素療法中止までの期間 など |
| 解析計画 |
臨床的改善は、事前に定義した7点順序尺度[スコア1:死亡、2:入院かつECMO又は侵襲的人工呼吸器による管理、3:入院かつ非侵襲的換気又は高流量酸素による管理、4:入院かつ低流量酸素による管理、5:入院しており、酸素吸入を要しないがSARS-CoV-2による感染症にかかわらず継続的な治療を要する、6:入院しており、酸素吸入及び継続的な治療は要しない(ただし、プロトコルに従った本剤の投与は除く)、7:退院]による評価において、ベースライン時から2点以上の改善が得られた場合と定義した。回復は、酸素吸入を要しない又は退院した場合と定義した。部分修正後の回復は、ベースラインスコア2~4から5~7への改善、あるいはベースラインスコア5から6又は7への改善として定義した。 主要評価項目は比例オッズモデルを用いて解析し、治療を独立変数として各ベクルリー(5日間又は10日間)群と標準療法群を比較した。比例オッズの仮定はScore testを用いて検証し、比例オッズの仮定が満たされなかった場合は、Wilcoxon rank sum testによるP値の裏付けを示した。第1種の過誤の確率をコントロールするために、ベクルリーによる治療効果の統計的有意性をBonferroni法に基づいて評価した。各仮説(ベクルリー5日間投与群対標準療法群、ベクルリー10日間投与群対標準療法群)は、αレベル0.025で検定した。 |
試験のサマリー
- レムデシビルの5日間投与は、中等症のCOVID-19患者に対して、標準療法と比較して臨床症状の改善に効果があることが示されました(主要評価項目、検証的な解析結果)。
- 試験デザイン
・GS-US-540-5774(SIMPLE-Moderate)試験は中等症のCOVID-19患者596名を対象とした無作為化非盲検並行群間試験であり、その結果はJAMAに公表されています。
- 無作為化後10日目に7点順序尺度で評価した臨床状態(主要評価項目、検証的な解析結果)
・レムデシビル5日間投与は標準療法と比較して有意に臨床的改善をもたらすことが示されました(オッズ比:1.65、95% CI 1.09-2.48;p=0.02、比例オッズモデル)。
・レムデシビル10日間投与と標準療法との間に有意差は示されませんでした(p=0.18、Wilcoxon rank sum test、検証的な解析結果)。
- 安全性(副次評価項目)
・有害事象はレムデシビル10日間投与群、5日間投与群、標準療法群でそれぞれ、59%、51%、47%に認められました。主な有害事象として、レムデシビル10日間投与群、5日間投与群、標準療法群で、悪心は9%、10%、3%、下痢は5%、6%、7%、低カリウム血症は7%、5%、2%、頭痛は5%、5%、3%に認められました。
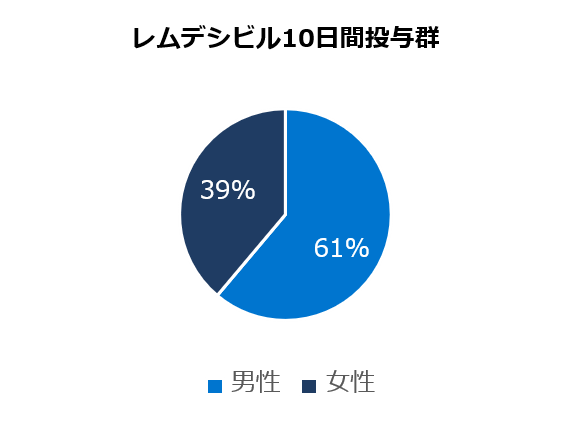
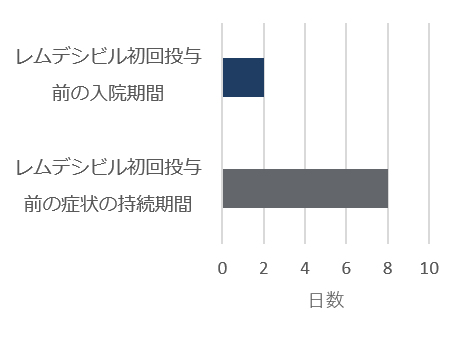
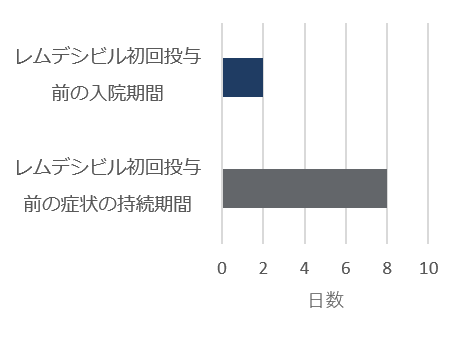
ベースライン時の人口統計学的及び臨床的特性








* アメリカンインディアン、アラスカ先住民、ハワイ先住民、太平洋諸島の民族、アラブ系民族、不明や特定不可能を含む。





* 医療が必要でなくても隔離やその他の社会的理由で入院が継続される場合も含まれる。

レムデシビル
10日間投与群
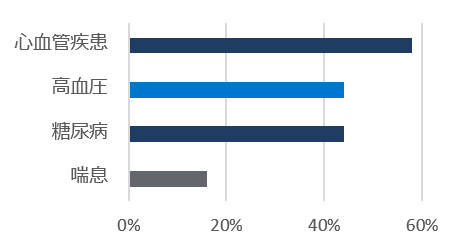
レムデシビル
5日間投与群

標準療法群
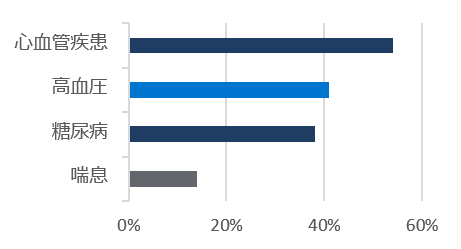

レムデシビル
10日間投与群
レムデシビル
5日間投与群
標準療法群

* レムデシビルの初回投与から最終投与の間(標準療法群では初日以降)に使用された薬剤を含む。
Spinner CD, et al.: JAMA 2020; 324(11): 1048-1057.
本試験はギリアド・サイエンシズ社より支援を受けています。著者にギリアド・サイエンシズ社より支援を受けている者が含まれます。
臨床転帰(主要評価項目・探索的評価項目)

a レムデシビル5日間投与群についてのオッズ比とP値は比例オッズモデルを用いて算出。レムデシビル10日間投与群については比例オッズ仮説が満たされていないためオッズ比は示されていない(P値はWilcoxon rank sum testで算出)。b 7点順序尺度においてベースラインから2点以上の改善。c ベースラインのスコア2~5から6~7、又はベースラインのスコア6から7への改善。
JAMA, 2020 Sep 15, 324(11):1048-1057, Copyright © 2020 American Medical Association. All rights reserved.
本試験はギリアド・サイエンシズ社より支援を受けています。著者にギリアド・サイエンシズ社より支援を受けている者が含まれます。
無作為化後10日目における臨床状態(主要評価項目)
- レムデシビル5日間投与は標準療法と比較して有意に臨床的改善をもたらすことが示されました(オッズ比:1.65、95% CI 1.09-2.48;p=0.02、比例オッズモデル、検証的な解析結果)。
- レムデシビル10日間投与と標準療法との間に有意差は示されませんでした(p=0.18、Wilcoxon rank sum test、検証的な解析結果)。

*:レムデシビル5日間投与群についてのオッズ比とP値は比例オッズモデルを用いて算出。レムデシビル10日間投与群については比例オッズ仮説が満たされていないためオッズ比は示されていない(P値はWilcoxon rank sum testで算出)。
- 無作為化後10日目の時点で退院となったのは、ベクルリー10日間投与群で65%、5日間投与群で70%、標準療法群で60%でした。

Spinner CD, et al.: JAMA 2020; 324(11): 1048-1057.より作図
本試験はギリアド・サイエンシズ社より支援を受けています。著者にギリアド・サイエンシズ社より支援を受けている者が含まれます。
有害事象の発現状況a(副次評価項目)
有害事象は5日間投与群で51%(98/191例)、10日間投与群で59%(113/193例)、標準療法群で47%(93/200例)に認められた。いずれかの群で5%を超えて発現した有害事象は悪心が5日間投与群で10%(19/191例)、10日間投与群で9%(18/193例)、標準療法群で3%(6/200例)、下痢が5日間投与群で6%(12/191例)、10日間投与群で5%(10/193例)、標準療法群で7%(14/200例)、低カリウム血症が5日間投与群で5%(10/191例)、10日間投与群で7%(13/193例)、標準療法群で2%(4/200例)、頭痛が5日間投与群で5%(10/191例)、10日間投与群で5%(10/193例)、標準療法群で3%(5/200例)であった。
重篤な有害事象は5日間投与群で9例(呼吸不全・呼吸困難・急性呼吸促迫症候群・脳血管障害・深部静脈血栓・発熱性好中球減少症・心拍数低下・治癒力低下・細菌性肺炎・肺塞栓症・腎仙痛各1件)、10日間投与群で10例(呼吸窮迫2件、急性呼吸不全・呼吸困難・ALT上昇・感染性関節炎・完全房室ブロック・意識レベルの低下・血行不安定・誤嚥性肺炎・失神・嘔吐各1件)、標準療法群で18例(急性呼吸不全5件、呼吸窮迫・呼吸不全・心停止各2件、急性腎障害・貧血・菌血症・癌性疼痛・胸痛・コロナウイルス感染症・輸液過負荷・腸管虚血・肺野陰影・心筋梗塞・肺炎各1件)に認められた。
投与中止に至った有害事象は、5日間投与群で4例(発疹2件、ALT上昇・心拍数低下各1件)、10日間投与群で8例(ALT上昇3件、AST上昇2件、高トランスアミナーゼ血症・血中アルカリホスファターゼ増加・血中ビリルビン増加・トランスアミナーゼ上昇・急性呼吸不全・低血圧各1件)に認められた。なお、標準療法群では投与中止と有害事象の関連を評価しなかった。
無作為化後27日目までの死亡は、5日間投与群で2例、10日間投与群で3例、標準療法群で4例に認められたが、いずれも本剤に関連する死亡ではなかった。

NA:not applicable、ULN:基準値上限、a:全安全性解析はデータカットオフ時までのすべての患者のデータを含む。b:28日間の試験期間を通じて
JAMA, 2020 Sep 15, 324(11):1048-1057, Copyright © 2020 American Medical Association. All rights reserved.
本試験はギリアド・サイエンシズ社より支援を受けています。著者にギリアド・サイエンシズ社より支援を受けている者が含まれます。
なお、これ以上の有害事象に関する詳細な情報は確認できないため、ベクルリー点滴静注用100mgの有害事象については電子添文をご参照ください。
